Welcome to iSARST
In this service, we implement two protein structural similarity search methods, SARST and CPSARST.
Besides, three outstanding structural alignment tools, FAST, TM-align and SAMO, are recruited as refinement engines. SE is applied to improve structure-based sequence alignments. We would like to thank these authors for their excellent developments, which greatly move this research field forward.
iSARST allows the user to input many structures at once. Its MPI system will do the similarity searches and structural alignments in a batch mode to rapidly retrieve structural homologs of the query proteins.
Subject Type
SARST is designed for co-linear structural similarity search while CPSARST specifically finds circular permutants.Circular permutation (CP) is a kind of structural rearrangement such that homologous proteins have different locations of termini. It is more and more widely used in protein engineering. Please visit CPDB for detailed information.


You can choose either SARST, the search engine for co-linear protein structural similarity search, or CPSARST, the search engine for circular permutants.
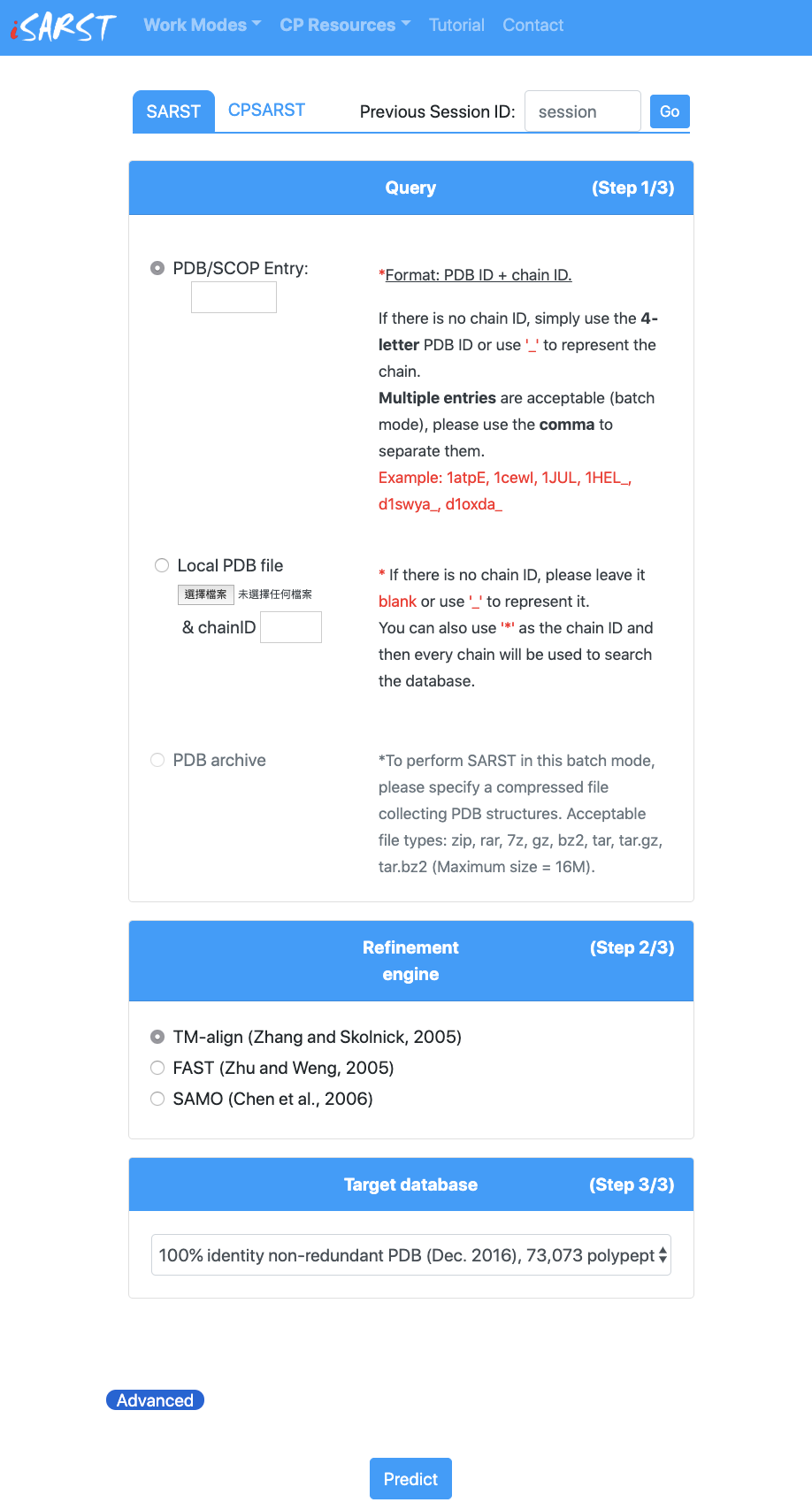
Input PDB entry/entries
There are several ways to make a request either by
-
entering PDB ID(s),
-
submitting PDB file or
-
uploading a compressed file.
1. Entering PDB ID(s)
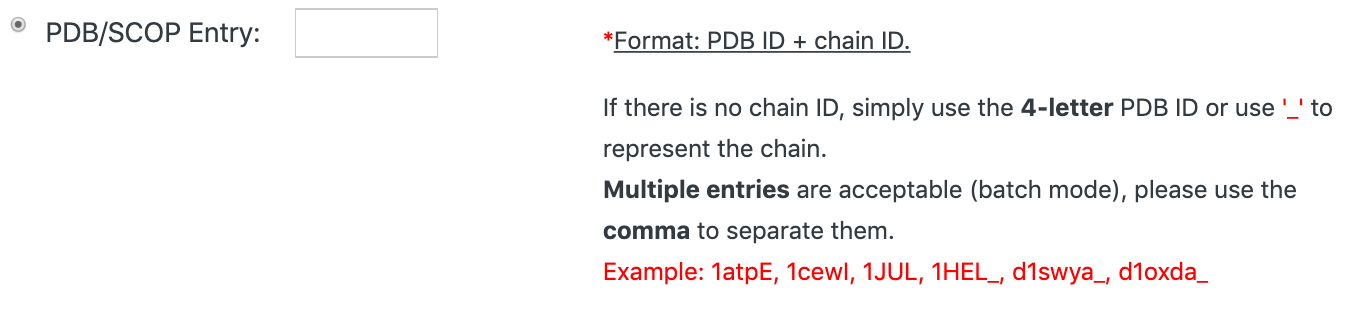
You can simply enter a PDB ID with the format: PDB ID+chain ID, to start your search.
If you have more than one query protein, please input all the PDB IDs at once with separating commas, like:
2. Submitting PDB file
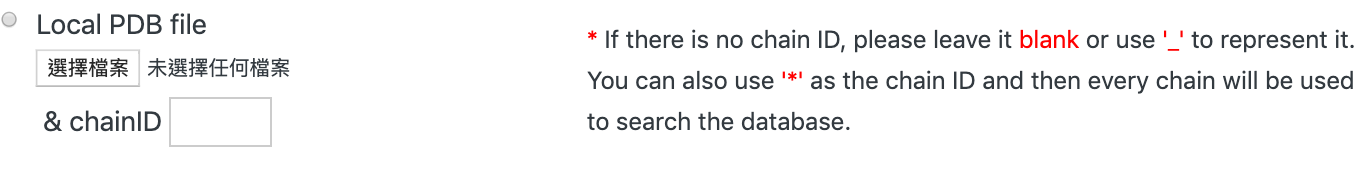
You can upload your local PDB file with its chain ID using the file field:
If you are not sure about the chain ID, or if you want to use every chain in the PDB structure to do the database search,
'*' or ' ' can be used as the chain ID.
' can be used as the chain ID.
3. Uploading a compressed file

Our Web service offers a convenient method to handle numerous PDB files. You can upload an archive file containing all your PDB structures. Currently iSARST accepts .zip .rar .tar.gz .tar formats. It will unzip your archive and search every chain in every protein simultaneously.
Retrieve Previous Results

After a submission, you will get a unique session ID. As the refinement calculations go on, you can close the browser and use the session ID to retrieve the results.
Provided that the cookies of your browser are enabled, the server will automatically list your previous session IDs when you return. Old sessions will be kept for 3 week in your browser. If you want to clear the acculated session IDs, simply click "Clear Sessions" and answer OK.
Setting
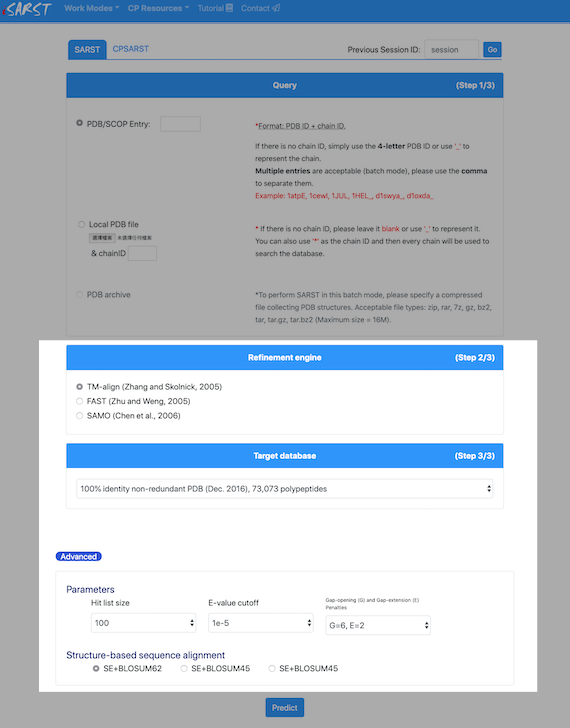
Target database
The whole PDB dataset as well as several non-redundantsubsets with various identity cut-offs are provided in iSARST.Parameters for search engines
You can adjust the size of hit list, E-value cut-off, andgap-panelties to optimize your search.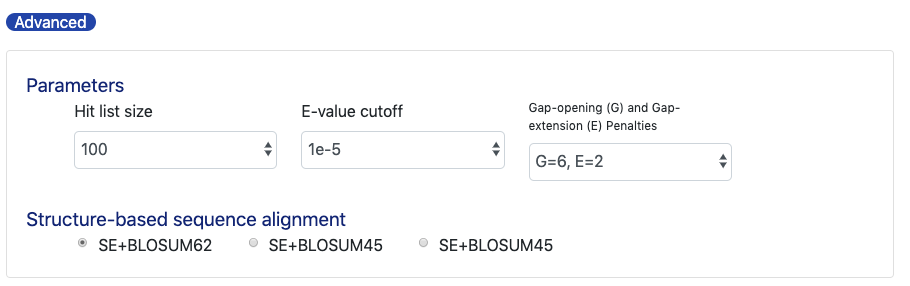
Refinement engine
The ordering of the hits retrieved by search engines will be further refined with accurate structural alignment tools. Currently we provide FAST, TM-align and SAMO as the refinement engines. The final hit lists will be ordered according to structural similarity scores calculated by them.Final Output
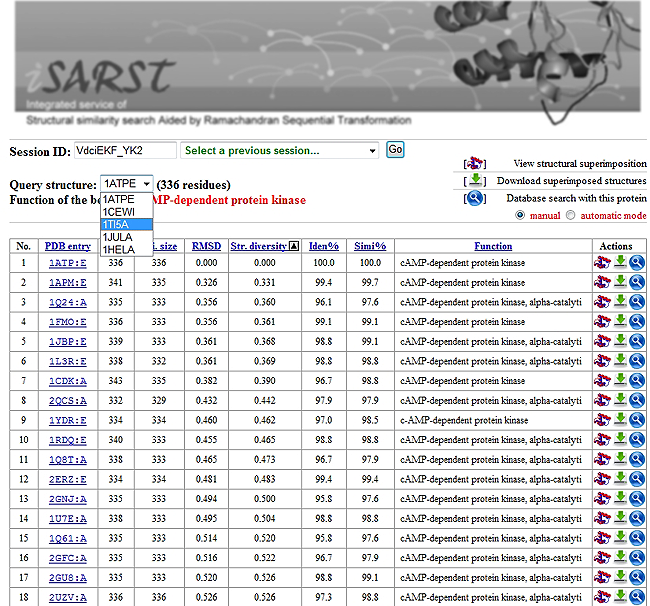
An example of the result page is shown in the right panel. If you submitted more than one protein, they will be listed in the selection menu.
The hit list can be re-ordered based on the PDB entry, protein size, align size, RMSD, structure diversity, sequence identities, sequence similarities and functions by clicking the column title. Functions of the 10 hits with the highest structural similarity score calculated by the selected refinement engine are highlighted in red to assist those who want to do a quick functional assignment.
Any protein in the hit list can be re-submitted to perform a new round of similarity search simply by clicking  . In the upper-right corner of this page, you can set the re-submission mode as manual or automatic. In the manual mode, you will be able to adjust the parameters of the search engine. In the automatic mode, a simple set of parameters will be used by iSARST. In the upper-right corner of this page, you can set the re-submission mode as manual or automatic. In the manual mode, you will be able to adjust the parameters of the search engine. In the automatic mode, a simple set of parameters will be used by iSARST.
. In the upper-right corner of this page, you can set the re-submission mode as manual or automatic. In the manual mode, you will be able to adjust the parameters of the search engine. In the automatic mode, a simple set of parameters will be used by iSARST. In the upper-right corner of this page, you can set the re-submission mode as manual or automatic. In the manual mode, you will be able to adjust the parameters of the search engine. In the automatic mode, a simple set of parameters will be used by iSARST.
Superimposed structures can be downloaded by clicking . To use our interactive on-line viewer (![]() ) properly, you need to download and install the MDLR® Chime plug-in.
) properly, you need to download and install the MDLR® Chime plug-in.
